
Haemoglobin Q-H disease (––/−αQ) is caused by the co-inheritance of Hb Q-Thailand and α0 thalassaemia. The anaemia is usually moderate, with a thalassaemia intermedia phenotype.
Hb Q-Thailand is caused by a point mutation in the α globin gene. Most heterozygotes for Hb Q-Thailand have the genotype (αα/−αQ), and have a thalassaemia trait phenotype.


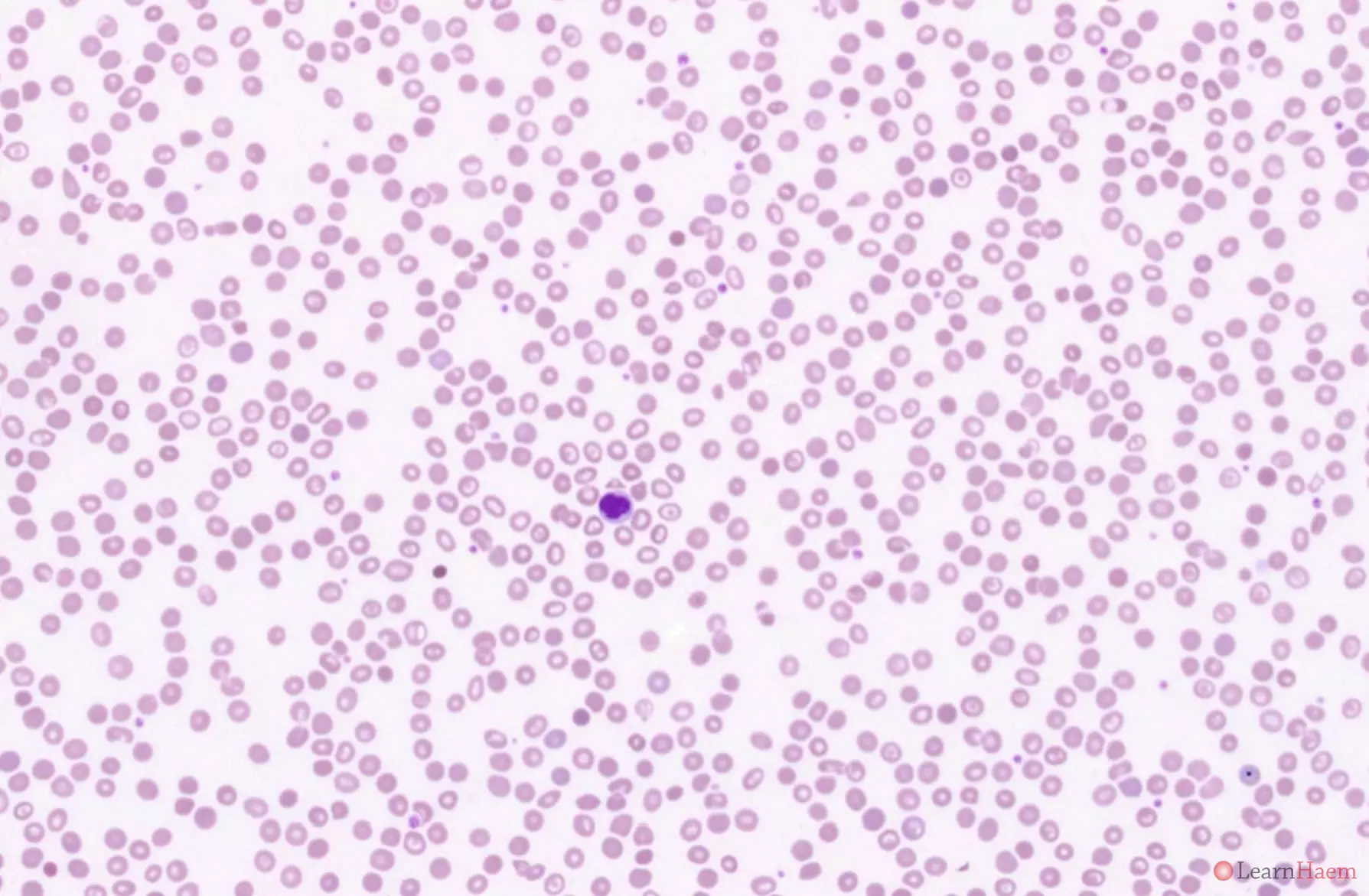
This is the blood film from a patient with Hb Q-H disease. This patient has numerous target cells.
Blood film features:
- The blood film looks similar to that of HbH disease
- Marked hypochromic, microcytic anaemia
- Anisopoikilocytosis with target cells, teardrop cells and fragments
- Basophilic stippling
- Polychromasia
Differential diagnosis:
- Iron deficiency anaemia (degree of anisopoikilocytosis is too severe for isolated iron deficiency).
- ß thalassaemia intermedia (such patients tend to have circulating nRBCs, which are absent in HbH disease. The reticulocytosis in HbH disease is also more prominent.).
- Hereditary pyropoikilocytosis (cells are normocytic and haemoglobinisation is normal).
- Congenital dyserythropoietic anaemias (often characterised by macrocytosis with no reticulocytosis).
- Acquired HbH disease (look for features of dysplasia, which may point towards an underlying myelodysplastic syndrome).
Haemoglobin electrophoresis:

Alkaline gel electrophoresis from a Patient with Haemoglobin Q-H Disease. There is a band with the mobility between that of haemoglobin F and haemoglobin S. Less well-depicted is a fast band corresponding to haemoglobin H.
The sickling test is negative in patients with haemoglobin Q.

Acid gel electrophoresis from a patient with haemoglobin Q-H Disease. It shows a variant haemoglobin with a mobility between Hb A and Hb S. This corresponds to Hb Q-Thailand. There is also a faint band with the same mobility of Hb A, likely representing Hb H.
HPLC:
HPLC from a patient with haemoglobin Q-H Disease. There is no normal Hb A as there are no normal α chains. There are two variant haemoglobins. The first has a very short retention time, and corresponds to haemoglobin H (red arrow). The second (blue arrow) has a retention time of 4.66 minutes, falling in the S window. This represents haemoglobin Q-Thailand. The findings differ from that of sickle cell anaemia in that Hb H is not seen in sickle cell anaemia, and the mobility on acid gel especially is not the same as Hb S.
Other resources:
- Clinical phenotype of haemoglobin Q-H disease
- PHE Family Origin Questionnaire
- PHE Sickle Cell and Thalassaemia Screening Programme (2019)
- BCSH Guideline: Significant Haemoglobinopathies: Guidelines for Screening and Diagnosis (2010)
- TIF Guideline: Management of Non-Transfusion Dependent Thalassaemia (2017)
- ASH Image Bank: HbH Disease
Leave A Comment