
ß thalassaemia can interact with haemoglobin variants, such as haemoglobin E and haemoglobin S, to produce clinically-significant phenotypes which range in severity from transfusion-dependent anaemias to chronic moderate anaemias.
ß thalassaemia mutations are common in the Mediterranean, India, Southeast Asia. They can occasionally be seen in Afro-Caribbeans.
Haemoglobin S arises from a point mutation in the ß globin gene. Sickle cell mutations are more common in individuals of African descent. They are also found in Central and South America, the Middle East and parts of India.
Patients with Hb S / ß thalassaemia have a sickle cell disease phenotype. The percentage of HbS may be offset by higher amounts of Hb F, Hb A2 and, in cases of ß+ thalassaemia, Hb A, but this is counterbalanced by the increased viscosity from a higher steady-state haemoglobin.


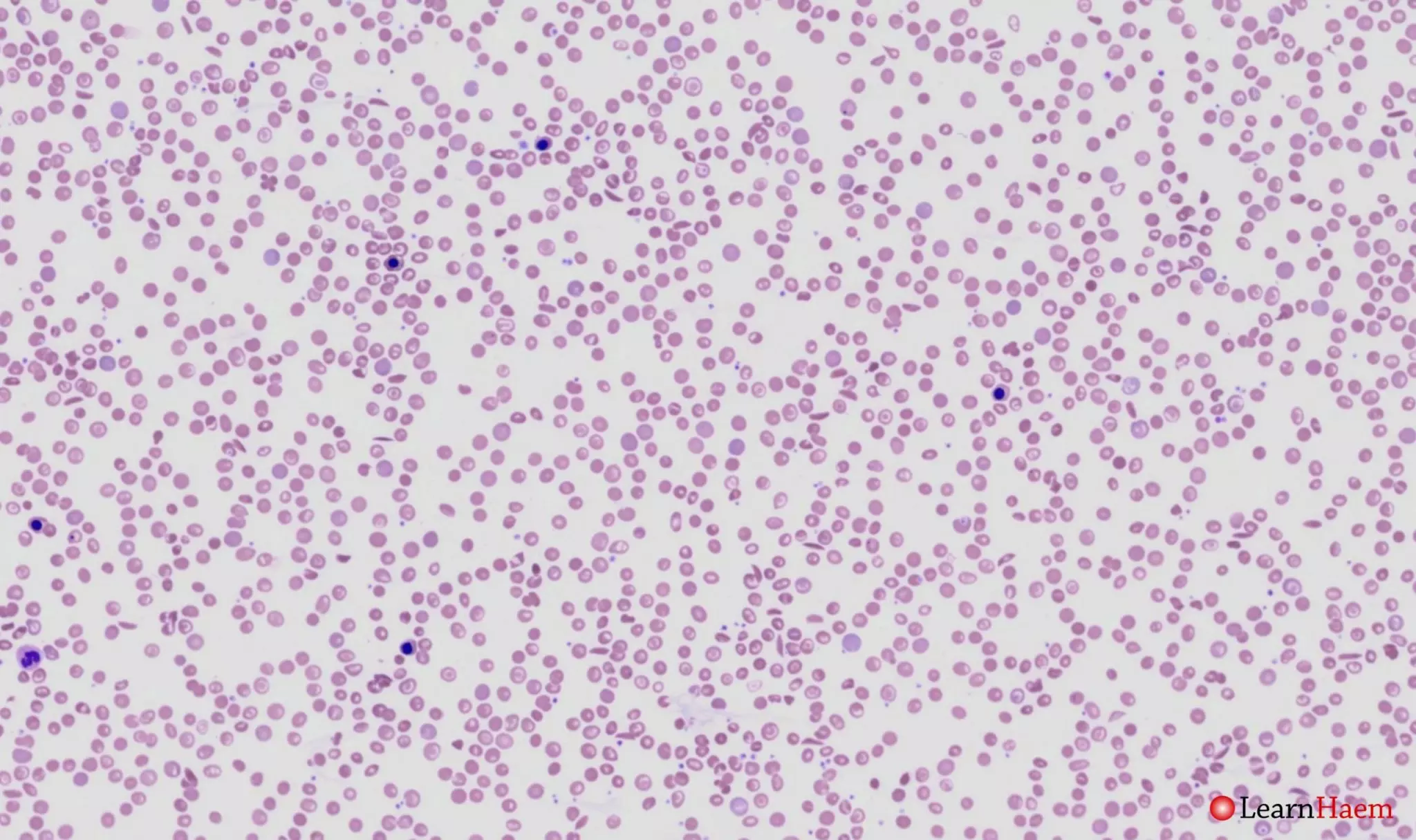
Blood film features:
- Severe microcytic, hypochromic anaemia
- Polychromasia
- Sickle cells
- Boat-shaped cells
- Target cells
- Basophilic stippling
- Numerous circulating nRBCs (some with defective haemoglobinisation)
- Features of hyposplenism
- Look for features of hydroxyurea therapy also
Differential diagnosis:
- Sickle cell anaemia (often less circulating nRBCs, basophilic stippling and other dyseythropoeitic features not prominent).
- Haemoglobin SC disease (will have characteristic SC poikilocytes, anaemia is less severe, less dyserythropoeitic features).
- ß thalassaemia intermedia / major (lacks sickle cells and boat-shaped cells). The sickle test in ß thalassaemia intermedia / major is negative.
Other investigations:
- Sickle test: positive in Hb S / ß thalassaemia
- Haemoglobin electrophoresis
- HPLC
Haemoglobin electrophoresis:

Alkaline gel electrophoresis from a patient with haemoglobin S / ß0 thalassaemia. There is no normal haemoglobin A. There are three bands. The most obvious has the mobility of haemoglobin S (red arrow). The differentials for this band are haemoglobins S, G, D and Lepore. There is a band with the mobility of haemoglobin F (green arrow), and another with the mobility of haemoglobin C (blue arrow). The differentials for such a band are haemoglobins C, E, O-Arab and A2.

Acid gel electrophoresis from a patient with haemoglobin S / ß0 thalassaemia. The dominant band has the mobility of haemoglobin S (red arrow). This is the same band that was seen on the alkaline gel, and excludes haemoglobin D and Lepore as differentials for that band. There is another band with the mobility of haemoglobin F (green arrow). The last band has the mobility of haemoglobin A (blue arrow), and corresponds to the band with the mobility of haemoglobin C on the alkaline gel. This excludes haemoglobin C and O-Arab as differentials. The possibilities for this band are therefore haemoglobins E and A2.
HPLC:
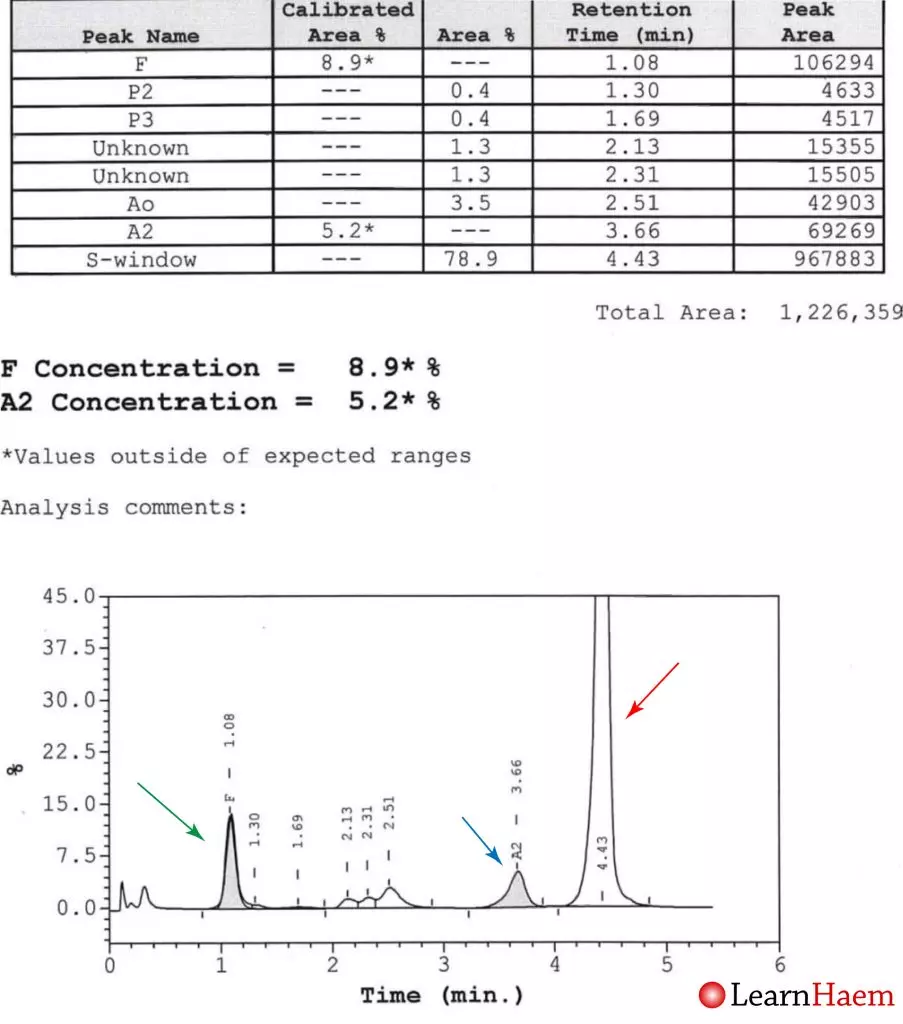
HPLC from a patient with haemoglobin S / ß0 thalassaemia. There is no normal haemoglobin A. There is an increased fraction of haemoglobin F (8.9%, normal adults have up to 1%). The major variant haemoglobin is in the S window (red arrow), excluding haemoglobin G-Philadelphia as a differential for the band with the mobility of haemoglobin S on electrophoresis. This is haemoglobin S. The other main fraction (5.2%) is in the A2 window (blue arrow). This fraction is too low to be haemoglobin E, and is hence haemoglobin A2. The Hb A2 fraction is higher than normal (up to 3.5% in normal individuals), meaning this patient like has a ß0 thalassaemia.
Other differentials for such an appearance include sickle cell anaemia (homozygous Hb S) with concomitant α thalassaemia (would not expect such a high Hb A2) or sickle cell anaemia with concomitant iron deficiency.
Other resources:
- PHE Family Origin Questionnaire
- PHE Sickle Cell and Thalassaemia Screening Programme (2019)
- BCSH Guideline: Significant Haemoglobinopathies: Guidelines for Screening and Diagnosis (2010)
- BCSH Guideline: Management of Acute Chest Syndrome in SCD (2015)
- BCSH Guideline: Red Cell Transfusion in SCD Part I (2016)
- BCSH Guideline: Red Cell Transfusion in SCD Part II (2016)
- BCSH Guideline: Use of hydroxycarbamide in children and adults with SCD (2018)
- BCSH Guideline: Red blood cell specifications for patients with hemoglobinopathies: a systematic review and guideline (2020)
- RCOG Guideline: Management of SCD in Pregnancy (2011)
Leave A Comment