
AE Bart’s disease is caused by the interaction of haemoglobin H disease and heterozygous haemoglobin E mutations. The clinical phenotype is of a moderate anaemia with severe microcytosis. Some patients may require intermittent transfusion support.


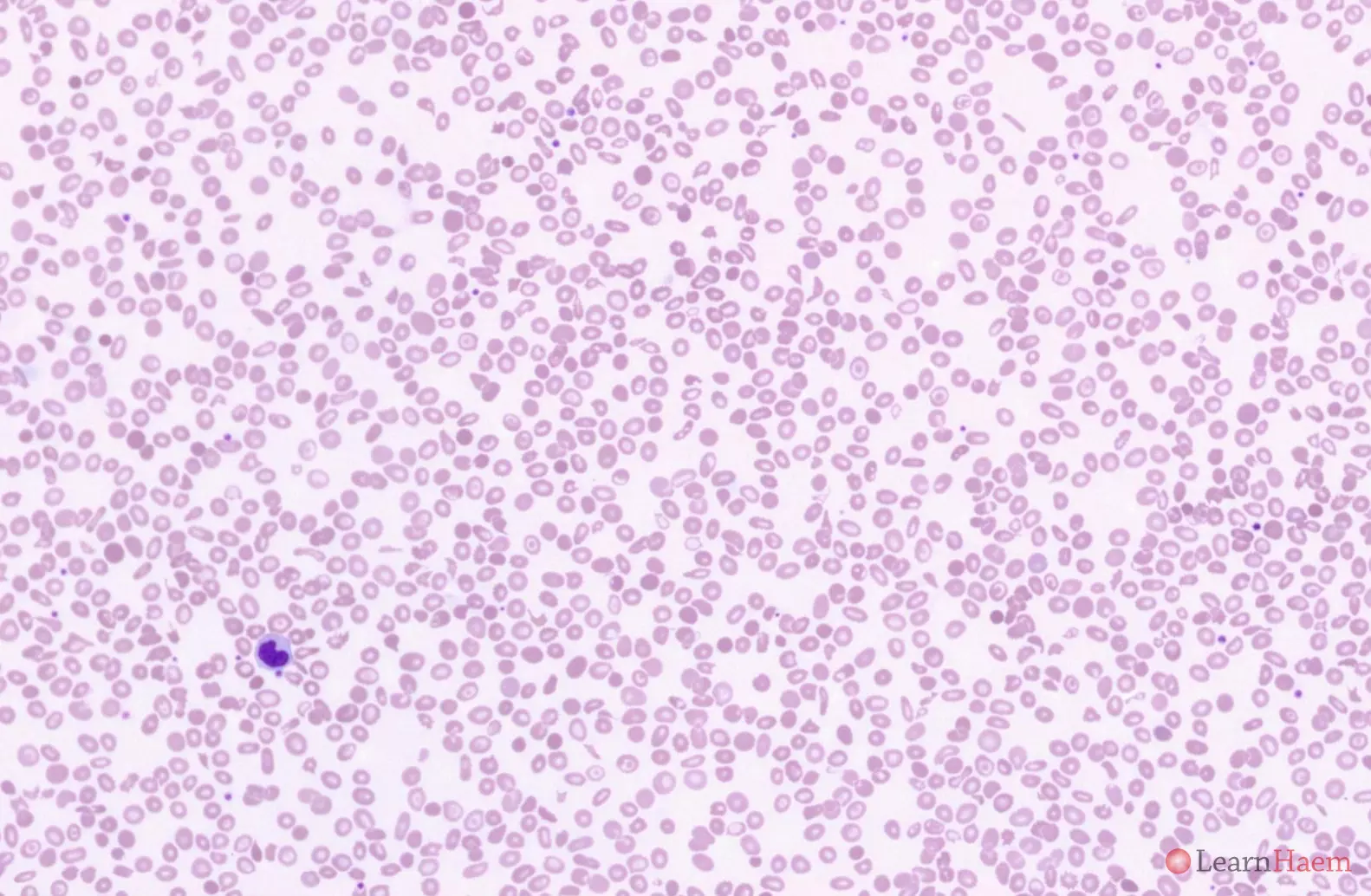
The blood film features are similar to that of HbH disease. There is marked anisopoikilocytosis. The Hb E heterozygous state is characterised by numerous target cells.
Haemoglobin electrophoresis:

Alkaline gel electrophoresis from a patient with AE Bart’s disease (lane 10). There is a normal band with the mobility of haemoglobin A. There are two variant haemoglobins. One is a fast band and corresponds to haemoglobin H (red arrow). The other has the same mobility of haemoglobin C (blue arrow), and likely represents haemoglobin E. The differentials for a such a band are haemoglobin C, E, O-arab and A2. The normal band at HbA means this patient has at least one normal ß globin gene.

Acid gel electrophoresis from a patient with AE Bart’s disease (black arrow). There is a single band with the mobility of haemoglobin A. This excludes haemoglobin C and O-Arab (mobility with Hb S on acid gel) as possible candidates for the band with the mobility of Hb C on alkaline electrophoresis.
HPLC:

HPLC from a patient with AE Bart’s disease. Normal haemoglobin A (70%) is present. There is a variant haemoglobin with a short retention time (red arrow), corresponding to haemoglobin H.
In addition, there is a variant haemoglobin in the A2 window (15%, blue arrow). The fraction is too high for beta thalassaemia intermedia (Hb A2 usually up to 10%). The usual fraction of haemoglobin E is ~30%. However, this patient has a concommitant alpha thalassaemia, which lowers the fraction of Hb E by about 5% for every deleted gene.
The percentage of haemoglobin F (green arrow), is higher than expected for a normal adult (6.5%, up to 1% in normal adults). This is likely to be a compensatory mechanism for the anaemia.
Other resources:
- Clinical and Molecular Features of AE Bart’s Disease
- PHE Family Origin Questionnaire
- PHE Sickle Cell and Thalassaemia Screening Programme (2019)
- BCSH Guideline: Significant Haemoglobinopathies: Guidelines for Screening and Diagnosis (2010)
- TIF Guideline: Management of Non-Transfusion Dependent Thalassaemia (2017)
- ASH Image Bank: HbH Disease
Leave A Comment