
Hereditary spherocytosis (HS) is an autosomal dominant condition characterised by mutations in red cell membrane proteins. The majority of mutations are in ankyrin and ß-spectrin genes, with a minority occuring in the red cell membrane protein, band 3 and α-spectrin genes. This results in a destabilised membrane and spherocytosis.
Clinical features:
- Phenotypic severity ranges from asymptomatic to severe chronic haemolysis.
- Most patients have compensated haemolysis with normal haemoglobin concentration but a reticulocytosis.
- Patients may also have an increased frequency of choledocholithiasis.
- Pregnancy may cause severe anaemia in non-splenectomised patients.
Diagnosis:
- In patients with a strong family history, typical blood film findings with a negative DCT are enough to confirm the diagnosis.
- Otherwise, the eosin-5-maleimide (EMA) binding test is the screening test of choice.
- EMA binds to transmembrane proteins.
- Fluorescence is measured via flow cytometry.
- HS results in a decrease in EMA binding and reduced fluorescence.


0 x
0 x
0 x
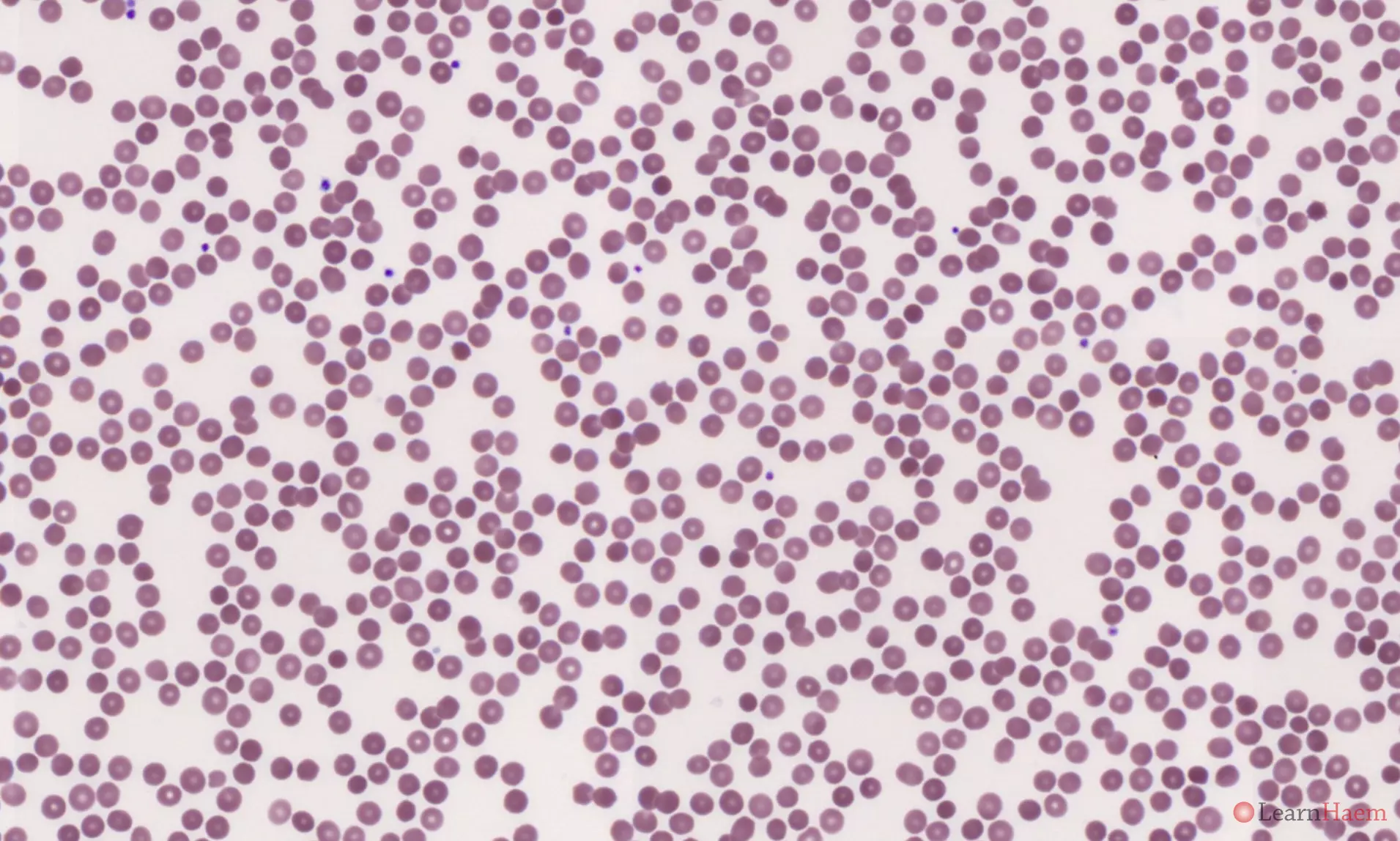
Blood film features:
- Anaemia is usually mild or absent
- Spherocytosis
- Polychromasia and polychromatic reticulocytes
0 x
0 x
Other features to look out for:
- Features of splenectomy:
- Howell-Jolly bodies
- Pappenheimer bodies
- Acanthocytes
- Target cells
- Spherocytes
- Stomatocytes
- Thrombocytosis
- Platelet anisocytosis
- Lymphocytosis
Treatment:
- Supportive measures:
- Folic acid supplementation.
- Transfusion support during haemolytic crises.
- Splenectomy is the treatment of choice for patients with severe HS.
- Splenectomy should ideally be delayed till after the age of 6.
- Patients should be vaccinated against encapsulated bacteria before splenectomy.
- Thromboprophylaxis should be given to all patients undergoing splenectomy.
Leave A Comment